Computational Cancer Genome Analysis
Team Leader
Projects
In the Computational Cancer Genome Analysis group, we analyze different genomic datasets (whole-exome and whole-genome sequencing, RNA-seq, DNA methylation data), and we develop innovative computational tools to better understand the origin and the molecular diversity of tumors. We explore several research axes, as described below.
Identification of genes driving hepatocellular carcinogenesis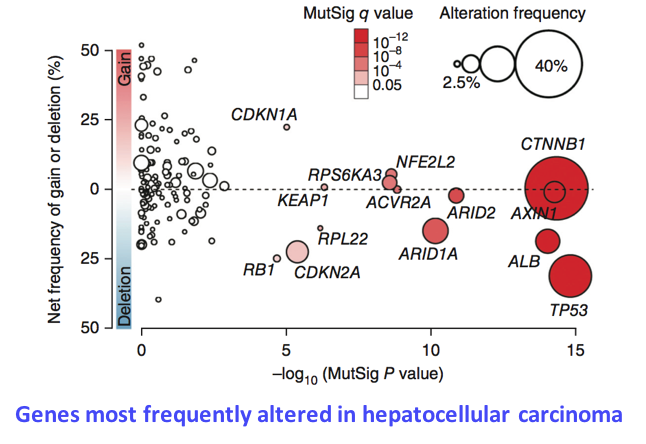
Tumor cells harbor numerous molecular alterations (mutations, chromosome gains and losses, translocations) that can alter the function or the activity of target genes. We develop computational approaches to integrate these alterations and identify genes recurrently altered, which are likely to play a key role in oncogenesis. By analyzing the exome sequences of 250 hepatocellular carcinomas, we recently identified 161 putative driver genes belonging to 11 major cellular pathways (Schulze, Imbeaud, Letouzé et al., Nat Genet 2015). We now analyze whole-genome sequencing data to identify non-coding mutations likely to affect the regulatory sequences of target genes, like the activating mutations of TERT promoter (Nault et al., Nat Commun 2013).
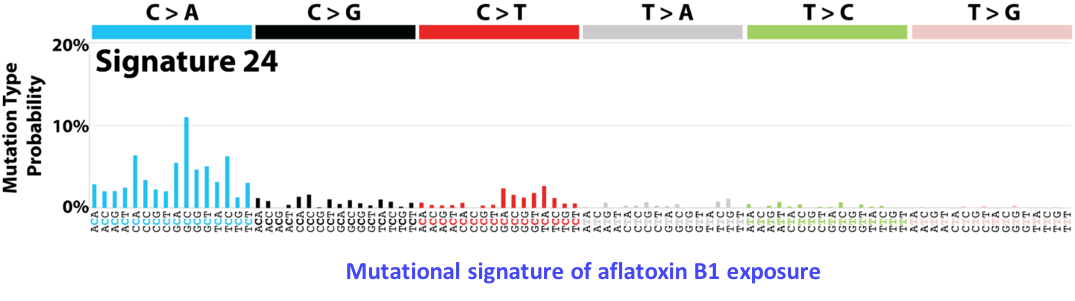
Mutational signatures
Somatic mutations that drive cancer progression are the consequence of spontaneous enzymatic conversions, replication errors, or mutagenic exposures like tobacco or UV light. These mutational processes leave imprints on the tumor genome that can be identified as mutational signatures, caracterized by specific types of mutations or mutations occurring in specific genomic contexts. For instance, tobacco carcinogenes induce mostly C>A mutations, whereas defects in DNA mismatch repair genes lead to an enriched frequency of C>T mutations in NCG trinucleotide context. By analyzing a large series of liver tumors by whole-exome sequencing, we have identified 2 new mutational signatures caracteristic of liver tumors (Schulze, Imbeaud, Letouzé et al., Nat Genet 2015). One of these signatures, caracterized by frequent C>A mutations at GCC trinucleotides, could be related to exposure to aflatoxin B1, a toxin produced by a mushroom in warm and wet countries of Africa and Asia. We now analyze whole-genome sequencing data from a new series of tumors, associated with diverse risk factors, to identify new signatures and distinguish mutational process operative in the early and late steps of oncogenesis. In collaboration with Pr Pierre Laurent-Puig’s team (UMR-S 1147), we also analyze the mutational signatures of other tumor types, like lung and colorectal cancer.
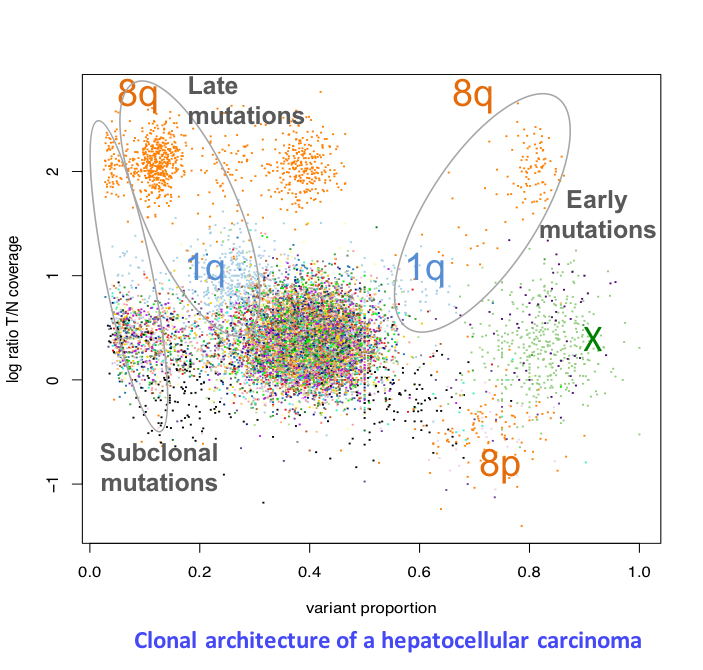
Clonal evolution of liver tumors
Tumors develop through the expansion of cell clones having acquired genomic alterations that confer them a proliferative advantage over surrounding cells. Several clones may coexist within a single tumor, including a dominant clone and one or more minor subclones. Understanding tumor heterogeneity is essential as subclones may harbor specific genetic defects conferring resistance to treatment. Besides, reconstructing the clonal architecture of a tumor allows to distinguish early from late genetic alterations, and to better understand the role of each driver gene. We currently analyze whole-genome sequencing data to reconstruct the clonal architecture of 50 liver tumors. In particular, we analyze cases of adenomas having progressed to carcinomas (Pilati et al., Cancer Cell 2014), to identify the molecular events triggering malignant transformation.
Epigenetic signatures of oncogenic processes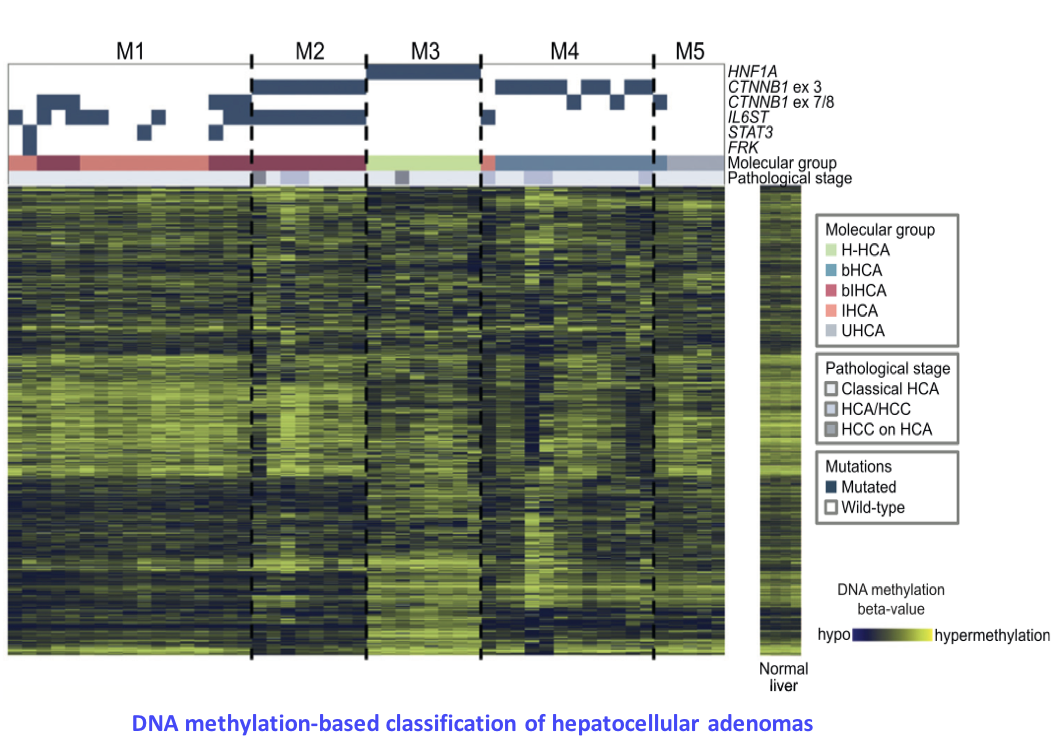
Epigenetic profiles (DNA methylation, histone modifications, chromatin conformation) are highly rearranged in tumor cells. Besides, numerous epigenetic regulators, implicated in DNA (de)methylation or chromatin remodeling, are frequently mutated, in particular in liver cancers. However, the mechanistic link between these molecular alterations and the epigenetic profiles of tumors remains poorly understood. By analyzing the methylome of a large series of liver tumors previously characterized by whole-exome sequencing, we wish to identify specific DNA methylation signatures. We will then correlate these signatures with clinical (exposure to risk factors) and molecular annotations (mutations in epigenetic regulators) to identify the cause of each pattern. This project aims at understanding how altered epigenetic profiles are established, and how the affect the transcriptome of tumor cells. In collaboration with INSERM UMR970 team (Dr Judith Favier), we also study the hypermethylator phenotype induced by succinate dehydrogenase mutations in paraganglioma (Letouzé et al., Cancer Cell 2013).
Insertional mutagenesis by AAV and HBV virus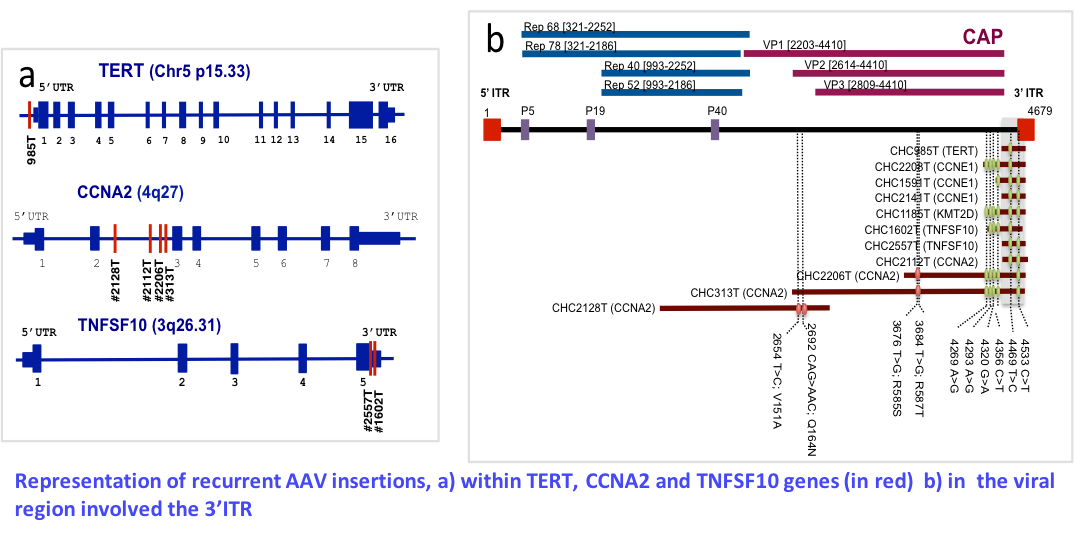
Virus, including hepatitis B virus (HBV) and hepatitis C, are major causes of hepatocellular carcinomas (HCC) worldwide. HBV is a well-known oncogenic DNA virus in liver tumors that induces insertional mutagenesis, chromosome instability and expression of oncogenic viral proteins. Currently only 2 other DNA viruses (merkel polyomavirus and human papilloma virus) are known to induce oncogenic insertional mutagenesis in human. Recently we obtained very innovative results showing that genomic approaches can help to identify new risk factors of HCC and modified our understanding of the disease. We showed that the adeno-associated virus type 2 (AAV2) is involved in HCC development on normal liver due to insertional mutagenesis in cancer genes (1). Currently, AAV is the 8th virus known associated to human cancer. The first identification of recurrent oncogenic AAV2 insertions in liver tumors leads us to develop a research project aiming to identify viral sequences and possibly new AAV integration sites in different types of liver tumors. As recombinant AAV is used as a vector for gene therapy in human clinical trials, a fine understanding of the mechanisms of carcinogenesis of the wild type virus is mandatory to assess the potential risk of cancer development after gene therapy. The objective is to quantify the contribution of AAV infection to the development of liver tumors in patients with liver disease related to various etiologies or without liver disease. We aim to identify new risk factors of hepatocellular carcinoma by analyzing tumor genomes and to validate them in epidemio-molecular studies through a comprehensive analysis of the tumor, host and viral genomes.
Transcriptome deregulation in liver tumors
Transcriptome profiling of liver tumors using DNA microarrays allowed us to identify homogeneous molecular groups of tumors, associated with distinct molecular alterations and risk factors (Boyault et al., Hepatology 2007). With the advent of high-throughput sequencing, we can now study the transcriptome by directly sequencing the RNAs extracted from a tumor sample (RNA-seq technique). These data give access to an unprecedented wealth of information. In addition to gene expression levels, RNA-seq data give access to the structure and sequence of transcripts, and allow the discovery of new genes. We have generated in the lab the RNA-seq profiles of a large series of liver tumors. We now develop innovative bioinformatic approaches to (1) refine the molecular classfication of liver tumors, (2) analyze the deregulation of non-coding RNAs, (3) detect alternative splicing and allele specific expression in tumors, and (4) identify the mutations that are actually expressed (more likely to play an oncogenic role) and RNA editing events.
Development of an annotation database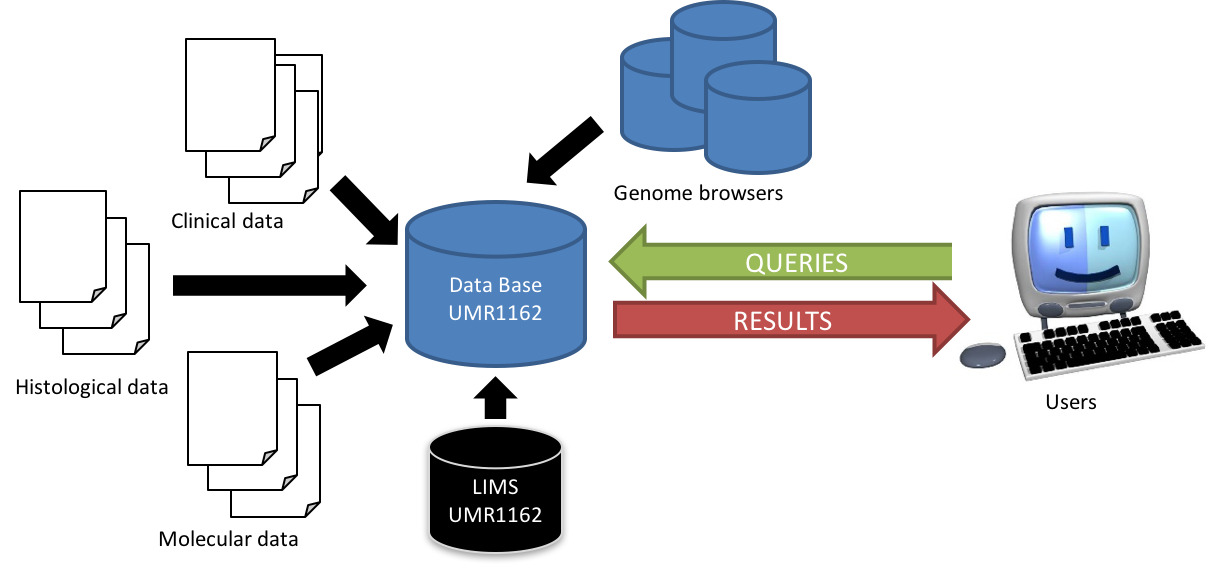
The development of genetics and new in-depth and high throughput technologies results in massive data production. Moreover, the multiplicity of programs and genomic annotations through “genome browsers” has created a “jungle” of terminology which requires the establishment and use of a unified vocabulary. In this context, we design and develop a database for structuring, annotation and exploitation of this mass of data generated by genomics programs. This project will, in collaboration with the EBCI Company, the implementation of an efficient IT infrastructure to meet our mixed data model, including clinic, histological and molecular datasets, to lead (1) the optimization of the architecture of our data model, (2) the management of the annotation including updates, and (3) optimizing a Web server to allow biologists to efficiently query data.
Team

University Paris 5
Publications
APC germline hepatoblastomas demonstrate cisplatin-induced intratumor tertiary lymphoid structures. Morcrette G, Hirsch TZ, Badour E, Pilet J, Caruso S, Calderaro J, Martin Y, Imbeaud S, Letouzé E, Rebouissou S, Branchereau S, Taque S, Chardot C, Guettier C, Scoazec JY, Fabre M, Brugières L, Zucman-Rossi J. Oncoimmunology. 2019 Mar 28;8(6):e1583547. doi: 10.1080/2162402X.2019.1583547. eCollection 2019.
Analysis of Liver Cancer Cell Lines Identifies Agents With Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Caruso S, Calatayud AL, Pilet J, La Bella T, Rekik S, Imbeaud S, Letouzé E, Meunier L, Bayard Q, Rohr-Udilova N, Péneau C, Grasl-Kraupp B, de Koning L, Ouine B, Bioulac-Sage P, Couchy G, Calderaro J, Nault JC, Zucman-Rossi J, Rebouissou S. Gastroenterology. 2019 May 4. pii: S0016-5085(19)36771-X. doi: 10.1053/j.gastro.2019.05.001.
A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, Imbeaud S, Bayard Q, Gustot T, Deviere J, Bioulac-Sage P, Calderaro J, Ganne-Carrié N, Laurent A, Blanc JF, Guyot E, Sutton A, Ziol M, Zucman-Rossi J, Nault JC. Hepatology. 2019 Mar 25. doi: 10.1002/hep.30623.
Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Bayard Q, Meunier L, Peneau C, Renault V, Shinde J, Nault JC, Mami I, Couchy G, Amaddeo G, Tubacher E, Bacq D, Meyer V, La Bella T, Debaillon-Vesque A, Bioulac-Sage P, Seror O, Blanc JF, Calderaro J, Deleuze JF, Imbeaud S, Zucman-Rossi J, Letouzé E. Nat Commun. 2018 Dec 7;9(1):5235. doi: 10.1038/s41467-018-07552-9.
PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Yang J, Trépo E, Nahon P, Cao Q, Moreno C, Letouzé E, Imbeaud S, Gustot T, Deviere J, Debette S, Amouyel P, Bioulac-Sage P, Calderaro J, Ganne-Carrié N, Laurent A, Blanc JF, Guyot E, Sutton A, Ziol M, Zucman-Rossi J, Nault JC. Int J Cancer. 2019 Feb 1;144(3):533-544. doi: 10.1002/ijc.31910. Epub 2018 Nov 9.
Systemic AA Amyloidosis Caused by Inflammatory Hepatocellular Adenoma. Calderaro J, Letouzé E, Bayard Q, Boulai A, Renault V, Deleuze JF, Bestard O, Franco D, Zafrani ES, Nault JC, Moutschen M, Zucman-Rossi J. N Engl J Med. 2018 Sep 20;379(12):1178-1180. doi: 10.1056/NEJMc1805673.
Palimpsest: an R package for studying mutational and structural variant signatures along clonal evolution in cancer. Shinde J, Bayard Q, Imbeaud S, Hirsch TZ, Liu F, Renault V, Zucman-Rossi J, Letouzé E. Bioinformatics. 2018 Oct 1;34(19):3380-3381. doi: 10.1093/bioinformatics/bty388.
Argininosuccinate synthase 1 and periportal gene expression in sonic hedgehog hepatocellular adenomas. Nault JC, Couchy G, Caruso S, Meunier L, Caruana L, Letouzé E, Rebouissou S, Paradis V, Calderaro J, Zucman-Rossi J. Hepatology. 2018 Sep;68(3):964-976. doi: 10.1002/hep.29884. Epub 2018 Jun 6.
Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Letouzé E, Shinde J, Renault V, Couchy G, Blanc JF, Tubacher E, Bayard Q, Bacq D, Meyer V, Semhoun J, Bioulac-Sage P, Prévôt S, Azoulay D, Paradis V, Imbeaud S, Deleuze JF, Zucman-Rossi J.